Was ist NF1?
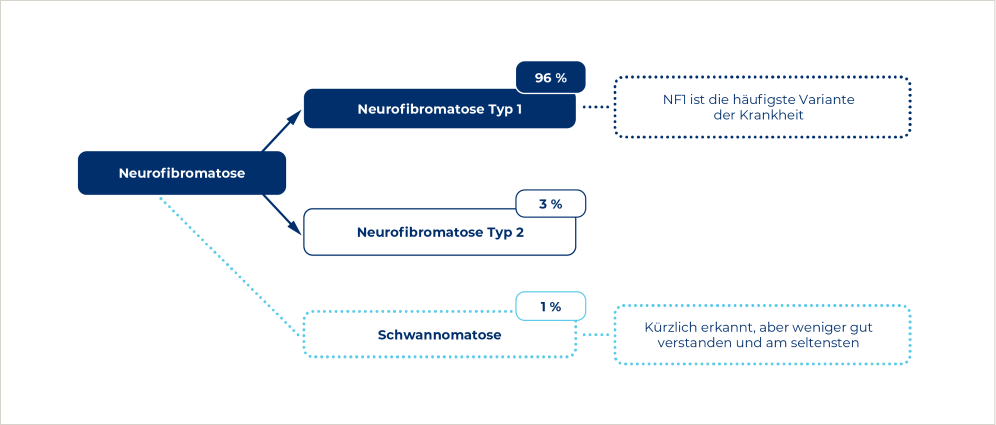
Neurofibromatose ist eine neurokutane Erkrankung, die von Tumoren des Nervensystems und der Haut gekennzeichnet ist.2 Die bei Weitem häufigste Form ist die Neurofibromatose Typ 1 (NF1) mit 96 %, gefolgt von Neurofibromatose Typ 2 (NF2) mit 3 % und Schwannomatose mit 1 %.3



Neurofibromatose Typ 1 assoziierte Plexiforme Neurofibrome (NF1-PN) und unerfüllter Bedarf
Erfahren Sie mehr über die Inzidenz von NF1-PN, die Belastung durch inoperable PNs und Behandlungsmöglichkeiten.
Obwohl NF1 eine seltene Krankheit ist, stellt sie eine relativ häufige neurogenetische Multisystemerkrankung dar, die bei geschätzt 1 von 3.000 bis 1 von 6.000 Neugeborenen auftritt.2 Sie ist mit kutanen, neurologischen, orthopädischen und neoplastischen Manifestationen verbunden, die zum Teil progressiv verlaufen und zu einer erheblichen Morbidität führen.2
NF1-Patienten haben ebenfalls ein erhöhtes Malignitätsrisiko und eine gegenüber der Allgemeinbevölkerung um rund 10-15 Jahre verkürzte Lebenserwartung.2

Neurofibromatose Typ 1 (NF1) im Fokus
Was versteht man unter Neurofibromatose Typ 1? In diesem Video erklärt Alex die Ursachen, Symptome und Behandlungsmöglichkeiten dieser Erkrankung.
Wodurch wird NF1 verursacht?
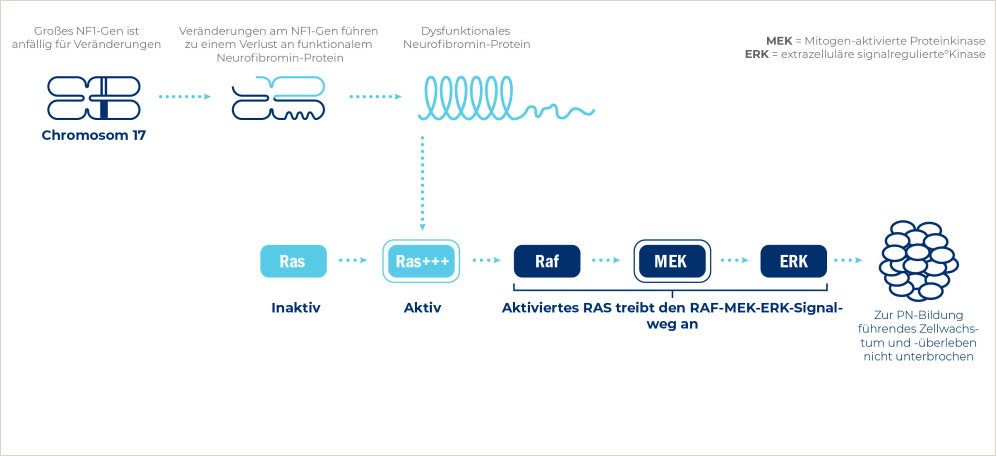
NF1 wird durch Mutationen im NF1-Gen verursacht, das für Neurofibromin codiert.4
Änderungen der Funktion von Neurofibromin, einem natürlichen Tumorsuppressorprotein, sind mit einer Überaktivierung von Ras verbunden, was zur Zellproliferation und im Endeffekt zur Tumorbildung führen kann.4,5
Was sind Plexiforme Neurofibrome (PN)?
Plexiforme Neurofibrome (PN) bei Neurofibromatose Typ 1 (NF1): Eine meist progressive Erkrankung, die erhebliche Auswirkung auf die Lebensqualität und das emotionale Wohlbefinden der Patienten haben kann1
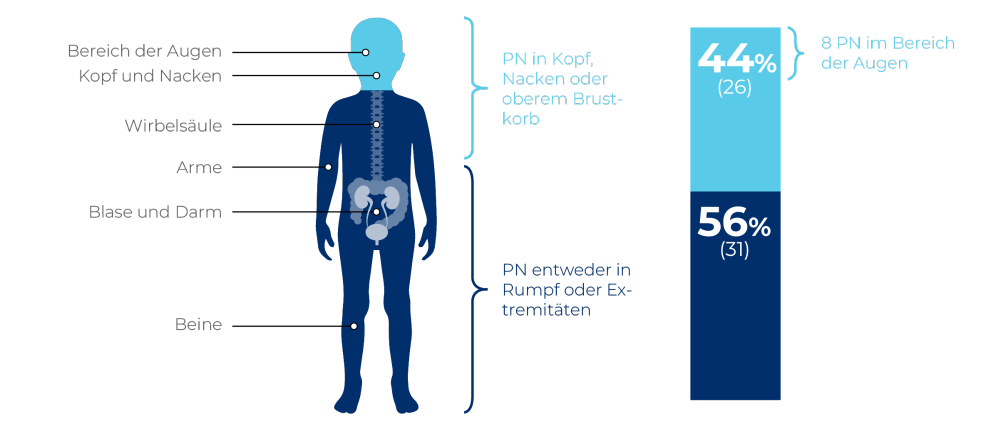
PN sind ein Merkmal der NF1 und gutartige Tumore, die sich im ganzen Körper entlang der Nervenscheiden entwickeln können.1,7 Auch wenn sie gutartig sind, so sind PN doch stark variabel und haben, je nach Größe und Lokalisierung, das Potenzial, schwerwiegende klinische Komplikationen hervorzurufen.4,7
Aufgrund der Unvorhersehbarkeit und der Komplexität der NF1 PN bringt die Behandlung dieser Krankheit besondere Herausforderungen mit sich.4,8
PN sind eine häufige Manifestation von NF1.9

Kommt es bei Plexiformen Neurofibromen zu einer Progression?
PN sind üblicherweise bei der Geburt bereits angelegt und zeigen gewöhnlich während der ersten zehn Lebensjahre ein rasches Wachstum.11 Einige PN sind unter Umständen so tief im Körper lokalisiert, dass sie unbemerkt bleiben, bis sich Schmerzen oder andere Symptome zeigen.12 Allerdings können PN schnell sehr groß werden.10,12
Unbehandelt kann die PN-Progression zu einer erheblichen Morbidität führen.1
Bei Patienten mit Plexiformen Neurofibromen kann es zu einem unkontrollierten und unvorhersehbaren Tumorwachstum kommen8,13
Die Daten der Natural History-Studie zeigen, dass es bei Kindern mit NF1 PN im Zeitverlauf zu einem unkontrollierten Tumorwachstum kommt.13
In einer Kohorte pädiatrischer Patienten (im Alter 3-18 Jahre) mit symptomatischen, inoperablen NF1 PN (N=93) zeigte sich bei keinem Patienten eine klinisch bedeutsame (≥20 %) Verringerung des PN-Volumens.14
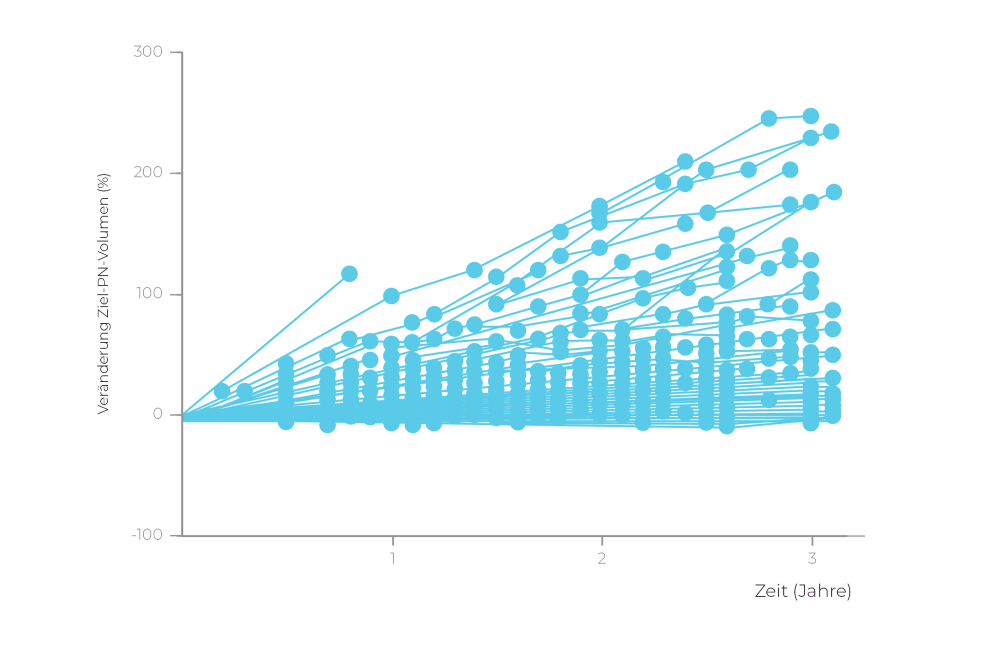
3 von 4 Patienten hatten eine Tumorprogression
(≥20 % PN-Volumenzunahme).‡1,14
PN sind hochgradig variabel und können überall im Körper entlang der Nerven wachsen.2,8,10
Es wurde gezeigt, dass Tumore mit einem Wachstum von >20 % pro Jahr wesentlich häufiger bei Kindern sind als bei Erwachsenen.15
PN können mit klinisch signifikanten Morbiditäten assoziiert sein, so dass eine frühzeitige Entdeckung und Intervention entscheidend ist.1
*Der Einschluss in die Natural History-Studie begann 2008 und dauert an1. Die Auswertungen der volumetrischen Kernspintomographie des NCI-Studienarms für pädiatrische Onkologie bis zum 15. Oktober 2018 wurden an AstraZeneca übermittelt, damit eine Analyse des PN-Wachstums und des progressionsfreien Überlebens der Patienten im Alter von 3 bis 18 Jahren erfolgen konnte, wobei die PN im Zusammenhang mit NF1 als externe Kontrollgruppe für die laufende SPRINT-Studie dienen, in der einige Patienten 5-7 Jahre behandelt werden.1
†92 Patienten werden in dieser Darstellung für die Natural History-Studie präsentiert. Die vollständige Natural History-Analysegruppe umfasst alle Patienten mit NF1-bezogenen PN (alle Altersgruppen), bei denen es mindestens 2 volumetrische MRI-Scans gibt1. Die altersangepasste Kohorte umfasst eine Untergruppe der vollständigen Natural History-Analysegruppe, einschließlich der Patienten mit mindestens 2 volumetrischen MRI-Scans, bei denen der erste Scan, der innerhalb der Altersgruppe 3-18 Jahre erfolgte, als Behandlungsbeginn betrachtet wurde1.
‡Die Mehrheit der PN wuchs stetig im Zeitverlauf oder blieb im besten Fall stabil (<20 % Volumenveränderung ab Behandlungsbeginn)1
Referenzen:
- Gross AM, Singh G, Akshintala S et al. Association of plexiform neurofibroma volume changes and development of clinical morbidities in neurofibromatosis 1. Neuro Oncol. 2018;20(12):1643–1651.
- Bergqvist C, Servy A, Valeyrie-Allanore L, et al. Neurofibromatosis 1 French national guidelines based on an extensive literature review since 1966. Orphanet J Rare Dis. 2020:15;37.
- Kresak JL and Walsh M. Neurofibromatosis: A Review of NF1, NF2, and Schwannomatosis. J Pediatr Genet. 2016;5(2):98–104.
- Hersh JH. Health supervision for children with neurofibromatosis. Pediatrics. 2008;121(3):633–642.
- Yap YS, McPherson JR, Ong CK, et al. The NF1 gene revisited—from bench to bedside. Oncotarget. 2014;5(15):5873–5892.
- Koselugo Fachinformation. Stand April 2022
- Dombi E, Ardern-Holmes SL, Babovic-Vuksanovic D, et al. Recommendations for imaging tumor response in neurofibromatosis clinical trials. Neurology. 2013;81(21 Suppl 1):S33–S40.
- Tonsgard JH. Clinical manifestations and management of neurofibromatosis type 1. Semin Pediatr Neurol. 2006;13(1):2–7.
- Jayachandran D, Sunantha S, Gopalaiah H, Veeraraghavan G. Plexiform neurofi bromatosis involving face and oral cavity. J Oral Maxillofac Pathol. 2014;18(1):114–117.
- Anderson JL. and Gutmann DH. Neurofibromatosis type 1. Handbook of Clinical Neurology Pediatric Neurology Part I. 2015;132:75–86.
- Hirbe AC and Gutmann DH. Neurofibromatosis type 1: A multidisciplinary approach to care. Lancet Neurol. 2014;13(8):834–843.
- Korf BR, Rubenstein AE. Neurofibromatosis: A Handbook for Patients, Families, and Health Care Professionals. 2nd Ed. Thieme Medical Publishers; 2005.
- Gross AM, Wolters PL, Dombi E, et al. Selumetinib in children with inoperable plexiform neurofibromas. N Engl J Med. 2020;382:1430–1442.
- Gross AM, Wolters PL, Dombi E, et al. Selumetinib in children with inoperable plexiform neurofibromas. N Engl J Med. 2020;382:1430–1442. Supplementary Appendix.
- Nguyen R, Dombi E, Widemann BC, et al. Growth dynamics of plexiform neurofibromas: a retrospective cohort study of 201 patients with neurofibromatosis 1. Orphanet J Rare Dis. 2012;7:75.