Introducing ULTOMIRIS® (ravulizumab)1
The first and only long-acting treatment for aHUS, providing1,3,4:
*Through 26 weeks; TMA response consisted of platelet count normalization, Serum LDH normalization and ≥25% improvement in serum creatinine from baseline
†54% (30/56; 95% CI: 40-67%) of adult and 77% (14/18; 95% CI: 52.4-93.6%) of pediatric patients met the composite endpoint of complete TMA response
‡Starting 2 weeks after the loading dose, maintenance doses are administered once every 4 or 8 weeks (depending on body weight)

*ULTOMIRIS® is administered as maintenance dose for patients above 20 kg every 8 weeks.
ULTOMIRIS® Mechanism of Action
With its extended half-life2, ULTOMIRIS® minimises the number of infusions
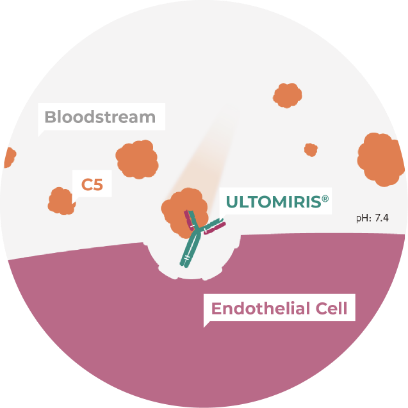
ULTOMIRIS® captures C5 in the bloodstream and prevents its activation2
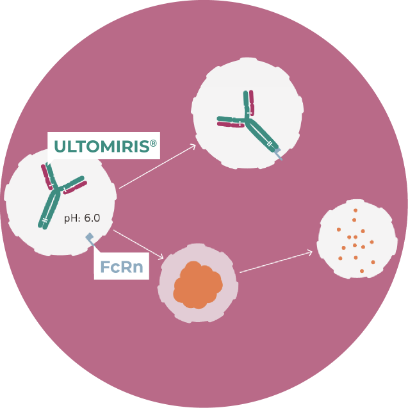
Specifically engineered, ULTOMIRIS® releases C5 in the acidified endosome, leaving C5 to be degraded by the lysosome. This acidified environment also increases the affinity of ULTOMIRIS® for the neonatal Fc receptor (FcRn), enhancing its return to the cell surface via FcRn-mediated recycling2
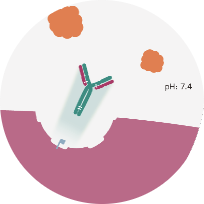
FcRn releases unbound ULTOMIRIS® back into the bloodstream to capture more C52
ULTOMIRIS® captures C5 in the bloodstream and prevents its activation2
Specifically engineered, ULTOMIRIS® releases C5 in the acidified endosome, leaving C5 to be degraded by the lysosome. This acidified environment also increases the affinity of ULTOMIRIS® for the neonatal Fc receptor (FcRn), enhancing its return to the cell surface via FcRn-mediated recycling2
FcRn releases unbound ULTOMIRIS® back into the bloodstream to capture more C52

Ravulizumab has a ~4x longer half-life than that of eculizumab®2§
§§Half-life of ULTOMIRIS® in patients with aHUS is 51.8 (16.2) days; mean elimination half life of eculizumab in aHUS is 12.4 days.
▼ Ultomiris (ravulizumab)
Selektivt immunsuppressivt middel, monoklonalt antistoff. ATC-nr.: L04A A43. Utleveringsgruppe C. Reseptbelagt legemiddel.
KONSENTRAT TIL INFUSJONSVÆSKE, oppløsning 300 mg/3 ml (100 mg/ml):
Indikasjoner: Behandling av pasienter med kroppsvekt ≥10 kg med atypisk hemolytisk uremisk syndrom (aHUS): Som er behandlingsnaive for komplementhemmer eller har fått ekulizumab i minst 3 måneder og har vist respons på ekulizumab. Behandling av voksne og barn ≥10 kg med paroksysmal nattlig hemoglobinuri (PNH): Hos pasienter med hemolyse med kliniske symptomer som indikerer høy sykdomsaktivitet. Hos pasienter som er klinisk stabile etter å ha vært behandlet med ekulizumab i minst de siste 6 månedene. Generalisert myasthenia gravis (gMG): Ultomiris er indisert som et tillegg til standardbehandling for behandling av voksne pasienter med gMG som er anti-acetylkolinreseptor (AChR) antistoffpositive. Dosering: Mht. sporbarhet skal preparatnavn og batchnr. noteres tydelig i pasientjournalen. Skal gis av helsepersonell og under tilsyn av lege med erfaring innen hematologiske sykdommer eller nyresykdommer. Voksne ≥18 år med PNH, aHUS eller gMG: Anbefalt dosering (basert på kroppsvekt) består av en startdose etterfulgt av vedlikeholdsdoser, se tabell 1 1 i preparatomtale (SPC). Vedlikeholdsdoser skal gis gis hver 8. uke, med oppstart 2 uker etter administrering av startdosen. Doseringsplanen kan unntaksvis avvikes fra med ± 7 dager for den planlagte infusjonsdagen, (unntatt for første vedlikeholdsdose av ravulizumab), men påfølgende dose skal administreres i henhold til opprinnelig plan. For pasienter som bytter fra ekulizumab til ravulizumab skal startdosen av ravulizumab gis 2 uker etter siste ekulizumabinfusjon, og deretter gis vedlikeholdsdoser hver 8. uke, med oppstart 2 uker etter administrering av startdosen, som vist i tabell 1. Hos gMGpasienter har behandling med ravulizumab kun blitt studert ved kronisk administrasjon. Ravulizumab har ikke blitt studert hos gMG-pasienter med MGFA klasse V. Barn og ungdom: ≥40 kg: Behandles iht. doseringsanbefalingene for voksne, se SPC. ≥10 kg til <40 kg: De vektbaserte dosene og doseringsintervallene er vist i tabell 3 i SPC. Spesielle pasientgrupper: Barn og ungdom: Sikkerhets- og effektdata av pasienter <10 kg er begrensede, ingen doseringsanbefalinger kan gis. Ravulizumab er ikke studert hos barn <30 kg med PNH og doseringen er fastsatt på grunnlag av data fra behandling av aHUS. Ravulizumab har ikke blitt studert hos pediatriske pasienter med gMG. Administrering: Fortynnet oppløsning skal kun gis som i.v. infusjon, gjennom et 0,2 μm-filter, vha. sprøytepumpe eller infusjonspumpe. Gis over en minimumsperiode på 0,17-1,3 timer (10-75 minutter), avhengig av kroppsvekt, se tabell 4 i SPC. Skal ikke gis som i.v. støt- eller bolusinjeksjon. Kontraindikasjoner: Overfølsomhet for innholdsstoffene. Aktiv Neisseria meningitidisinfeksjon ved behandlingsstart. Pasienter som ikke nylig er vaksinert mot Neisseria meningitidis, med mindre de får profylaktisk behandling med relevante antibiotika frem til 2 uker etter vaksinasjon. Forsiktighetsregler: Alvorlig meningokokkinfeksjon: Ravulizumab gjør pasienten mer utsatt for meningokokkinfeksjon/-sepsis (Neisseria meningitidis) pga. virkningsmekanismen. Meningokokksykdom forårsaket av enhver serogruppe kan oppstå. For å redusere infeksjonsrisikoen må alle pasienter vaksineres mot meningokokkinfeksjon minst 2 uker før behandlingsoppstart, med mindre risikoen ved å utsette behandling oppveier risikoen for å få meningokokkinfeksjon. Tilfeller av alvorlig meningokokkinfeksjon/-sepsis er rapportert hos ravulizumabpasienter. Tilfeller av alvorlig eller fatal meningokokkinfeksjon/-sepsis er rapportert ved bruk av andre terminale komplement-hemmere. Alle pasienter skal overvåkes for tidlige tegn på meningokokkinfeksjon og -sepsis, utredes umiddelbart ved mistanke om infeksjon og behandles med relevante antibiotika. Pasienten skal informeres om slike tegn/symptomer og at legehjelp skal søkes umiddelbart. Legen skal gi pasienten informasjonsbrosjyre og pasientkort. Immunisering: Før behandlingsoppstart anbefales det at pasienter starter med immunisering iht. gjeldende retningslinjer for immunisering. Vaksinasjon kan aktivere komplement ytterligere. Som følge av dette kan pasienter med komplement-medierte sykdommer oppleve økte tegn/symptomer på underliggende sykdom. Pasienten skal derfor overvåkes nøye for sykdomssymptomer etter anbefalt vaksinasjon. Pasienter <18 år skal vaksineres mot Haemophilus influenzae og pneumokokkinfeksjoner, og de nasjonale vaksinasjonsanbefalingene for den enkelte aldersgruppe skal følges nøye. Andre systemiske infeksjoner: Ravulizumab skal gis med forsiktighet ved aktiv systemisk infeksjon. Pasienten skal få informasjon i pakningsvedlegget for å øke oppmerksomheten rundt mulige alvorlige infeksjoner og tegn/symptomer. Legen skal gi pasienten råd om forebygging av gonoré. Infusjonsreaksjoner: Infusjonsreaksjoner og allergiske- eller overfølsomhetsreaksjoner (inkl. anafylaksi) kan oppstå ved ravulizumabinfusjon. Ved infusjonsreaksjon skal infusjonen avbrytes og nødvendige støttetiltak iverksettes ved tegn på kardiovaskulær ustabilitet eller respirasjonshemming. Seponering ved PNH, aHUS og gMG: Se Dosering i SPC. Hjelpestoffer: Inneholder 0,18 g natrium pr. 72 ml ved maks. dose (tilsv. 9,1% av WHOs anbefalte maks. daglige natriuminntak for voksne) etter fortynning med NaCl 0,9% injeksjonsvæske. Interaksjoner: For utfyllende informasjon om relevante interaksjoner, bruk interaksjonsanalyse på felleskatalogen. Ingen interaksjonsstudier er utført. Se SPC for veiledning i tilfelle samtidig behandling med intravenøs immunoglobulin, plasmabytte og plasmaferese Graviditet, amming og fertilitet: Fertile kvinner bør bruke sikker prevensjon under og i opptil 8 måneder etter behandling. Hos gravide kan bruk overveies etter nytte-/risikovurdering. Amming: Risiko for spedbarn som ammes kan ikke utelukkes. Bivirkninger: Svært vanlige (kan ramme flere enn 1 av 10 personer): hodepine diaré, kvalme, magesmerter, feber (pyreksi), tretthet (fatigue), øvre luftveisinfeksjon, forkjølelse (nasofaryngitt), ryggsmerter, leddsmerter (artralgi) Vanlige (kan ramme opptil 1 av 10 personer): svimmelhet, oppkast, fordøyelsesbesvær etter måltider (dyspepsi), elveblest, utslett, kløe i huden (pruritus), muskelsmerter (myalgi) og muskelspasmer, influensaliknende sykdom, frysninger, svakhet (asteni), infusjonsrelatert reaksjon, allergisk reaksjon (overfølsomhet), Urinveisinfeksjon Mindre vanlige (kan ramme opptil 1 av 100 personer): meningokokkinfeksjon, alvorlig allergisk reaksjon som medfører pustevansker eller svimmelhet (anafylaktisk reaksjon), gonokokkinfeksjon. Oppbevaring og holdbarhet: Oppbevares i kjøleskap (2-8°C), og i ytteremballasjen for å beskytte mot lys. Skal ikke fryses. Etter fortynning: Skal brukes umiddelbart. Kjemisk og fysikalsk stabilitet av fortynnet oppløsning er imidlertid vist i opptil 24 timer ved 2-8°C eller i opptil 4 timer ved romtemperatur. Andre opplysninger: Ultomiris rekvireres via CustomerOperationsEU@Alexion.com Pakninger og priser: Anbud: nei. Blå resept: nei. Byttbar: nei. Basert på SPC godkjent av SLV/EMA: 21.09.2022. Innehaver av markedsføringstillatelsen: Alexion Europe SAS, 103-105 rue Anatole France, 92300 Levallois-Perret, Frankrike. Repr.: Alexion Pharma Nordics AB, Stockholm, Sverige. alexion.nordics@alexion.com. Sist endret: 13-07-2023 Les felleskatalogtekst eller preparatomtalen (SPC) for mer informasjon, se www.felleskatalogen.no