LAL-D
(Deficiencia de lipasa ácida lisosomal)
La deficiencia de lipasa ácida lisosomal (LAL-D) es una enfermedad rara, rápidamente progresiva y potencialmente mortal(1,2)
La edad de inicio es muy variable, pudiéndose presentar en lactantes, niños y adultos con complicaciones metabólicas que pueden ser graves(1,3)
Los lactantes presentan la progresión más rápida resultando en muerte generalmente dentro de los primeros 6-12 meses de vida(1,3)
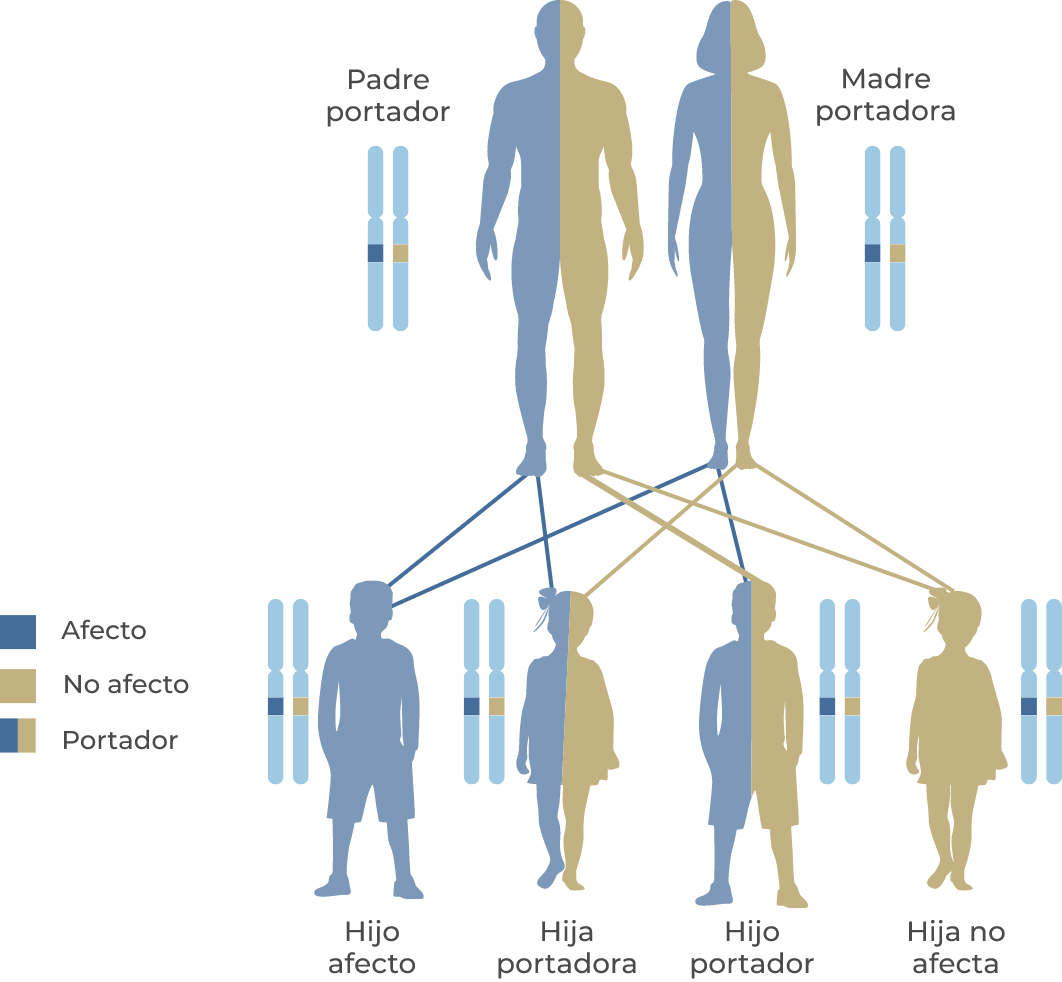
LAL-D es una enfermedad autosómica recesiva causada por mutaciones en el gen LIPA que interrumpen la actividad de la lipasa ácida lisosomal (LAL)(2,4)

Creado a partir de Gulani A y Weiler T, 2022.(5)
LAL-D
Podemos diferenciar 3 puntos importantes en la deficiencia de lipasa ácida lisosomal:
MECANISMO YPRESENTACIÓN CLÍNICA CLAVES EN EL DIAGNÓSTICO ABORDAJE TERAPÉUTICO
LAL: lipasa ácida lisosomal; LAL-D: deficiencia de lipasa ácida lisosomal
LIPA: lipasa A, tipo ácido lisosomal.
MECANISMO DE ENFERMEDAD
La enzima LAL es esencial para la hidrólisis de los ésteres de colesterol y triglicéridos en los lisosomas(1)
Como consecuencia de la deficiencia de LAL, los lípidos se acumulan progresivamente en el interior de los lisosomas, lo que conduce a la disfunción celular en múltiples órganos y tejidos vitales, como el hígado o el bazo, entre otros órganos(1,2)
Diferencias a nivel de homeostasis del colesterol celular en individuos sanos y en pacientes con LAL-D

Figura adaptada de Reiner Z, et al. 2014.(2)
¿Quieres saber más sobre la acumulación lisosomal de EC y TG en LAL-D?
SABER MÁSACAT: acyl-CoA cholesterol acyltransferase; cLDL: colesterol LDL; CVLDL: colesterol VLDL; EC: ésteres de colesterol; FA: ácido graso; FC: colesterol libre; FFA: ácido graso libre; HMG-CoA r: 3-hidroxi-3-metil-glutaril-CoA reductasa; LAL: lipasa ácida lisosomal; LAL-D: deficiencia de lipasa ácida lisosomal; LDL: lipoproteína de baja densidad; LDLR: receptor de LDL; SREBPs: proteínas de unión a elementos reguladores de esteroles; TG: triglicéridos; VLDL: lipoproteína de muy baja densidad.
SIGNOS Y SÍNTOMAS
LAL-D es una enfermedad heterogénea, con signos y síntomas de progresión muy variable entre los individuos afectados(2)
Signos y síntomas clínicos
Hepáticos

- Fibrosis y cirrosis
- Hepatomegalia / hepatoesplenomegalia
- Esteatosis microvesicular
- Insuficiencia hepática
- Disfunción de la vesícula biliar
- Colestasis
- Esplenomegalia
La hepatomegalia y esplenomegalia se han observado en el 99 % y el 74 % de niños y adultos con LAL-D, respectivamente (3)
Gastrointestinales

- Vómitos
- Diarrea
- Esteatorrea
- Distensión abdominal
- Dolor abdominal y epigástrico
- Malabsorción
- Varices esofágicas
Cardiovasculares

- Aneurisma
- Enfermedad de las arterias coronarias
- Dislipidemia
- Accidente cerebrovascular
Otros
- Trastornos de crecimiento*
- Calcificaciones de las glándulas suprarrenales*
- Anemia
- Caquexia
Marcadores séricos
Hepáticos
- Niveles elevados de transaminasas séricas (ALT, AST)
Lipídicos
- Niveles elevados de colesterol total
- Incremento del colesterol LDL
- Disminución del colesterol HDL
- Niveles elevados de triglicéridos
Tabla adaptada de Reiner Z, et al. 2014.(2)
ALT: alanina aminotransferasa; AST: aspartato transaminasa; HDL: lipoprotína de alta densidad; LAL-D: deficiencia de lipasa ácida lisosomal; LDL: lipoproteína de baja densidad; TG: triglicéridos.
*Relacionados con LAL-D RP
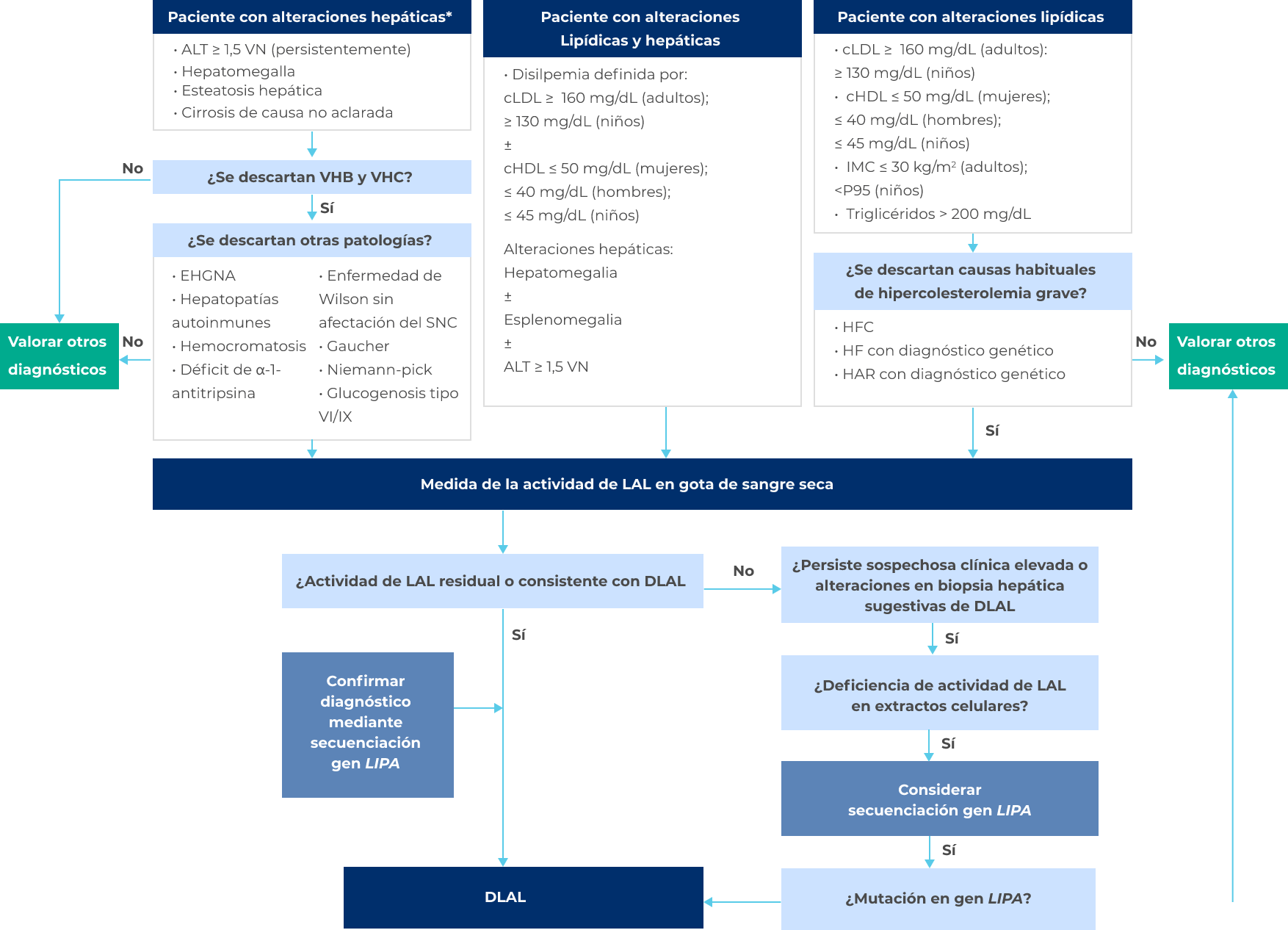
CLAVES EN EL DIAGNÓSTICO
Los pacientes con LAL-D se presentan dentro de un espectro, con edad de inicio y gravedad de la enfermedad variables(1,2,3-6,7,8)
Formas de presentación
- Fallo de medro
- Síntomas digestivos (vómitos, diarrea, síndrome de malabsorción)
- Organomegalias: mayoritariamente esplenomegalia
- Anemia
- Posible evolución a fallo hepático
- Calcificación de las glándulas suprarrenales
- Afectación hepática: hepatopatía crónica, hepatomegalia, colestasis, hipertensión portal
- Alteración lipídica: dislipemia, enfermedad aterosclerótica
- Otras afectaciones: esplenomegalia, diarrea, síndrome de malabsorción, hiperparatiroidismo
- Posible evolución a fallo multiorgánico
LAL: lipasa ácida lisosomal; LAL-D: deficiencia de lipasa ácida lisosomal.
ABORDAJE TERAPÉUTICO
Durante años, el único tratamiento disponible para el manejo del LAL-D ha consistido en medidas de soporte centradas en reducir las complicaciones de la enfermedad.(2,9)
Las opciones terapéuticas actuales incluyen:(9)
Tratamiento hipolipemiante:
La introducción de dietas bajas en grasas y la administración de estatinas han demostrado efectos beneficiosos en pacientes con LAL-D, como la reducción de la síntesis endógena de colesterol. Sin embargo, existe evidencia sobre la progresión del daño hepático a largo plazo a pesar del tratamiento.(9)
Trasplantes:
-
Médula ósea: En lactantes con LAL-D rápidamente progresivo, el trasplante de células madre hematopoyéticas puede considerarse como opción de tratamiento, si bien es necesario tener en cuenta la alta morbimortalidad asociada.(9)
-
Hígado: El trasplante hepático es el tratamiento actual para pacientes que presentan cirrosis hepática descompensada. Sin embargo, la intervención no trata la causa subyacente de la enfermedad que puede progresar en el corazón y los riñones. Además, la evidencia clínica es limitada y variable, lo que dificulta la obtención de conclusiones sobre la eficacia del tratamiento.(2.3)
Tratamiento de sustitución enzimática:
El objetivo del tratamiento de sustitución enzimática es tratar la causa subyacente de la enfermedad con la restauración la actividad de LAL, evitando así la progresión del LAL-D.(9,11)
La aprobación de los tratamientos basados en el reemplazo enzimático ha supuesto un gran avance terapéutico en el control de la enfermedad, pues diversos estudios han demostrado la eficacia y seguridad del tratamiento temprano con  Kanuma (sebelipasa alfa) en pacientes con LAL-D.(9)
Kanuma (sebelipasa alfa) en pacientes con LAL-D.(9)
LAL: lipasa ácida lisosomal.
Este medicamento está sujeto a seguimiento adicional, es prioritaria la notificación de sospechas de reacciones adversas asociadas a este medicamento.
Para acceder a la Ficha Técnica de Kanuma®, haz click aquí.
PRESENTACIÓN Y PRECIO: KANUMA 2 MG/ML concentrado para solución para perfusión, 1 vial. C.N: 709943. PVL notificado: 8.980 €. Financiado por el Sistema Nacional de Salud.
RÉGIMEN DE PRESCRIPCIÓN Y DISPENSACIÓN: Medicamento sujeto a prescripción médica. Uso Hospitalario. Consulte la Ficha Técnica completa antes de prescribir este medicamento.
REFERENCIAS
- Jones SA, Valayannopoulos V, Schneider E, et al. Rapid progression and mortality of lysosomal acid lipase deficiency presenting in infants. Genet Med. 2016;18(5):452-458.
- Reiner Ž, Guardamagna O, Nair D, et al. Lysosomal acid lipase deficiency--an under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis. 2014;235(1):21-30.
- Bernstein DL, Hülkova H, Bialer MG, et al. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol. 2013;58(6):1230-1243.
- Del Angel G, Hutchinson AT, Jain NK, et al. Large-scale functional LIPA variant characterization to improve birth prevalence estimates of lysosomal acid lipase deficiency. Hum Mutat. 2019;40(11):2007-2020.
- Gulani A, Weiler T. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022. Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK546620/. Acceso: noviembre 2022.
- Witeck CDR, Schmitz AC, de Oliveira JMD, et al. Lysosomal acid lipase deficiency in pediatric patients: a scoping review. J Pediatr (Rio J). 2022;98(1):4-14.
- Burton BK, Silliman N, Marulkar S. Progression of liver disease in children and adults with lysosomal acid lipase deficiency. Curr Med Res Opin. 2017;33(7):1211-1214.
- Burton BK, Deegan PB, Enns GM, et al. Clinical Features of Lysosomal Acid Lipase Deficiency. J Pediatr Gastroenterol Nutr. 2015;61(6):619-625.
- Camarena C, Aldamiz-Echevarria LJ, Polo B, et al. Update on lysosomal acid lipase deficiency: Diagnosis, treatment and patient management. Med Clin (Barc). 2017;148(9):429.e421-429.e410.
- Muntoni S, Wiebusch H, Jansen-Rust M, et al. Prevalence of cholesteryl ester storage disease. Arterioscler Thromb Vasc Biol. 2007;27(8):1866-1868.
- Kohli R, Ratziu V, Fiel MI, et al. Initial assessment and ongoing monitoring of lysosomal acid lipase deficiency in children and adults: Consensus recommendations from an international collaborative working group. Mol Genet Metab. 2020;129(2):59-66.
- Hamilton J, Jones I, Srivastava R, et al. A new method for the measurement of lysosomal acid lipase in dried blood spots using the inhibitor Lalistat 2. Clin Chim Acta. 2012;413(15-16):1207-1210.
- Li W and Lee MS. Dried Blood Spot. Wiley Online Library. Online ISBN:9781118890837. 2014.
- Ficha Técnica Kanuma®. Alexion Europe SAS. Versión Junio 2023.
- Burton BK, Balwani M, Feillet F, et al. A Phase 3 Trial of Sebelipase Alfa in Lysosomal Acid Lipase Deficiency. N Engl J Med. 2015;373(11):1010-1020.
- Burton BK, Feillet F, Furuya KN, et al. Sebelipase alfa in children and adults with lysosomal acid lipase deficiency: Final results of the ARISE study. J Hepatol. 2022;76(3):577-587.
ES/KAN-L/0013 Mayo 2024