¿Qué es la NF1?
La neurofibromatosis tipo 1 (NF1) es una enfermedad neurocutánea multisistémica genética rara e incurable, clínicamente heterogénea y con una evolución impredecible . Así los pacientes con NF1 además de presentar manifestaciones cutáneas, oftalmológicas y óseas, tienen una alta predisposición a desarrollar tumores en el sistema nervioso central y periférico3
Afecta a 1 de cada 3.000 personas y es uno de los trastornos genéticos raros más comunes3
El 97% de los casos de NF1 se diagnostica principalmente antes de los 8 años3
La NF1 es una enfermedad crónica y compleja que limita la calidad de vida de las personas que la padecen2
¿Qué causa la NF1?
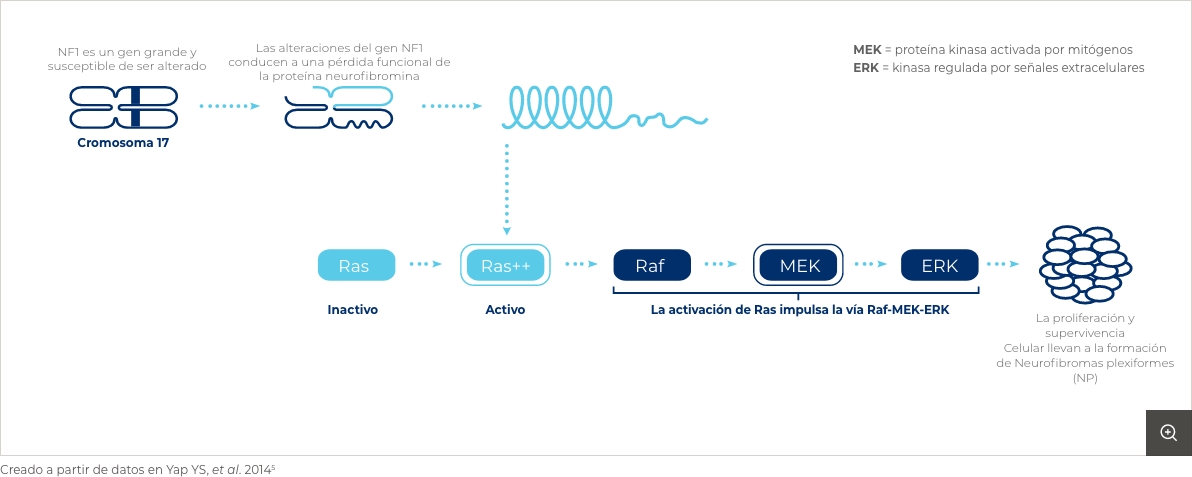
La NF1 se produce como consecuencia de una mutación en el gen NF1 . Este gen codifica para una proteína (neurofibromina) que actúa como supresor tumoral. Al estar mutado, se forma una proteína anormal que no puede ejercer su función, por lo que aumenta el riesgo de proliferación celular. Estas mutaciones pueden heredarse (en el 50% de los casos se trata de una enfermedad autosómica dominante) u ocurrir de manera espontánea (50% de los casos)4,5
Criterios de diagnóstico de la NF1
El diagnóstico de la NF1 consiste en la presencia de 2 o más de las siguientes manifestaciones. En el caso de que uno de los progenitores del paciente padeciera la enfermedad, solo tendría que cumplir uno de los criterios siguientes para ser diagnosticado.
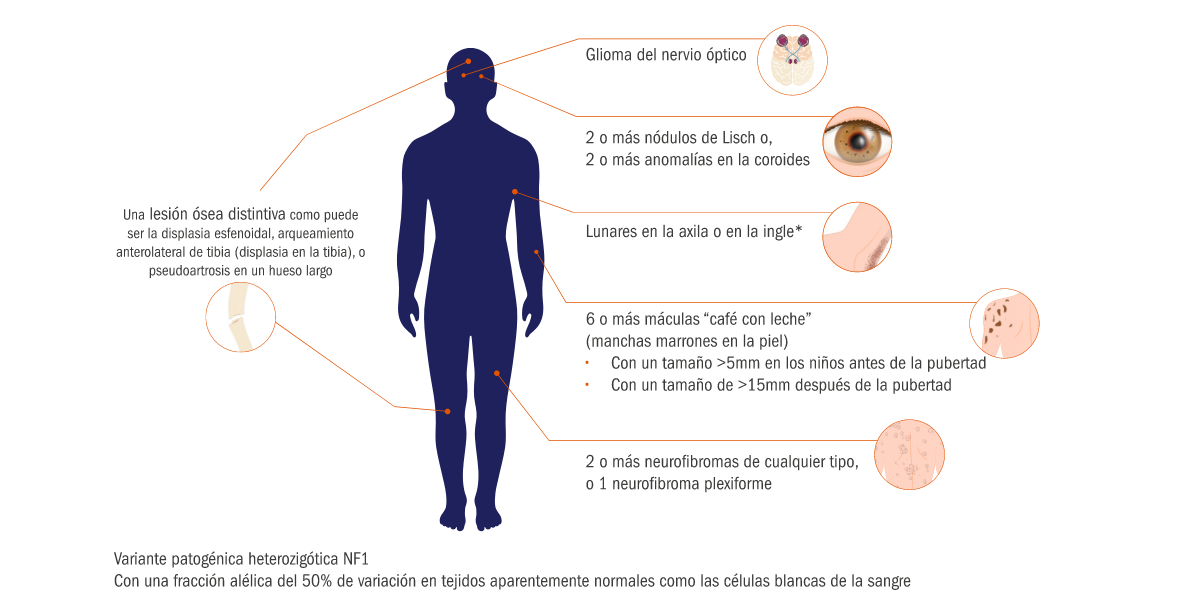
*Por lo menos una de las dos alteraciones pigmentarias (máculas “café con leche” o lunares) debería ser bilateral. Adaptado de CTF. Diagnostic criteria for Neurofibromatosis type 1 (NF1)6
de los pacientes con NF1 puede desarrollar Neurofibromas plexiformes (NPs)8,9
Los NPs son tumores benignos complejos, grandes, deformantes e invasivos, que involucran nervio, músculo, tejido conectivo, elementos vasculares y piel10, y por lo tanto afectan a la salud física, el bienestar emocional y el desarrollo social de los pacientes.
¿Son progresivos los NPs?
Los tumores plexiformes suelen estar presentes desde el nacimiento y pueden desarrollarse a lo largo de la infancia, adolescencia e incluso en la edad adulta temprana4,11
Los pacientes con un NPs pueden manifestar una forma sintomática del tumor.12 A medida que los NPs crecen, pueden causar morbilidades severas (ej. dolor, desfiguración y disfunción motora, problemas intestinales y de vejiga).13 Estas manifestaciones afectarían no solo a las actividades diarias sociales del paciente, sino también a su funcionalidad física y estado emocional de ansiedad y tristeza. Asimismo, hay un gran impacto en el bienestar emocional y la vida cotidiana de sus familias y cuidadores.14,15 Por lo tanto, la detección e intervención tempranas son críticas.1
Conoce más sobre los NPs
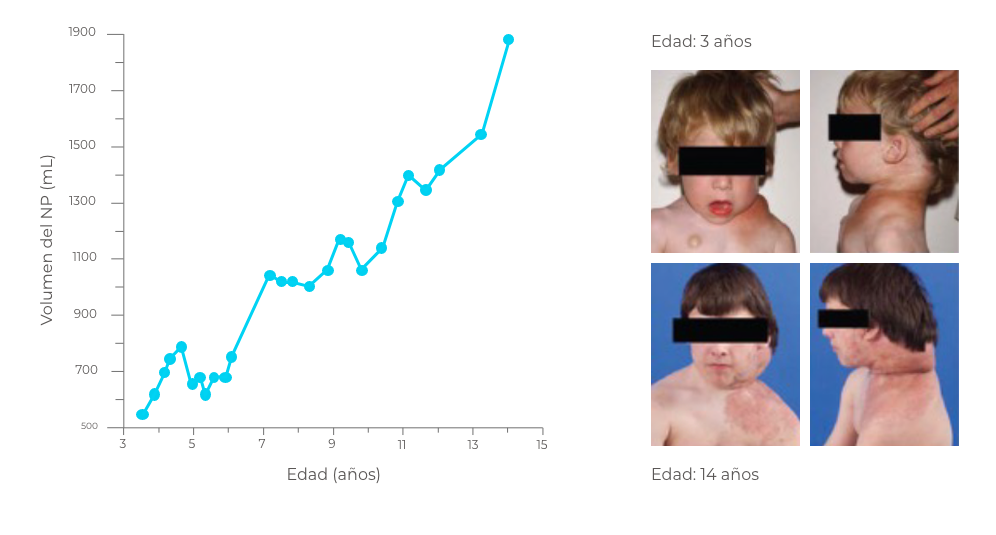
El crecimiento del NPs es impredecible (entre el 2,8 y el 15,9% al año) e imposible de controlar.1
Las tasas de crecimiento más rápidas de NPs se observan en pacientes de 3 a 5 años, con una tasa de crecimiento promedio del 35% al año.1
Creado a partir de datos de Gross AM, et al. 20181
La variabilidad y la naturaleza progresiva de la NF1, hace que sea difícil determinar el pronóstico tras el diagnóstico, especialmente a una edad temprana.17
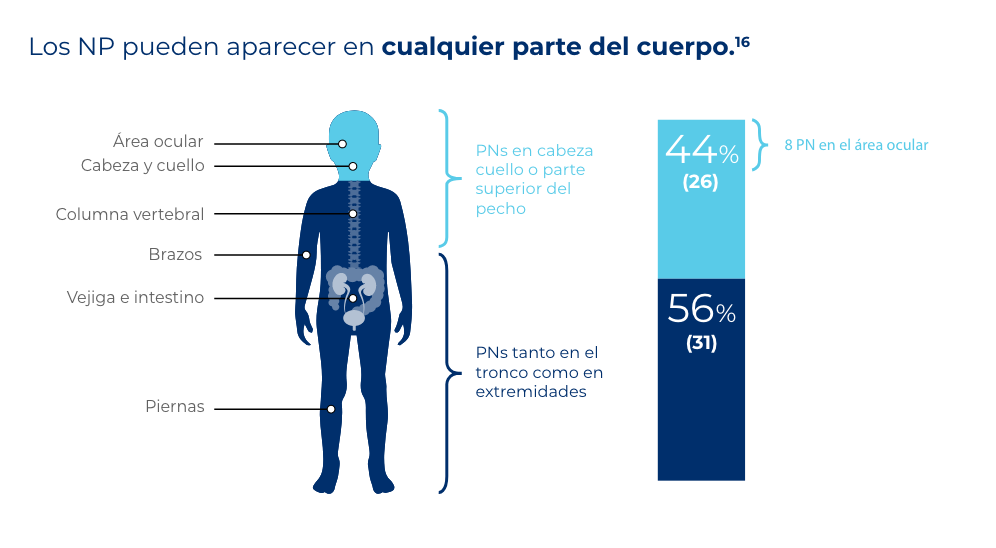
La aparición de los NPs en algunas partes como la cabeza y el cuello pueden ser señal de complicaciones clínicas graves (desfiguración facial y déficits funcionales).16
Adaptado de Gross AM, et al. 20181
Entre un 8-13% de los pacientes con NPs pueden presentar riesgo de desarrollar un tumor maligno de la vaina del nervio periférico (MPNST, por sus siglas en inglés).15
Los MPNST se asocian con dolor severo, progresión rápida de la enfermedad, baja respuesta al tratamiento y mal pronóstico. Son la causa principal de muerte en la NF1.18


